April 15, 2011
| Automatic Structure Verification (ASV)
This entry was e-mailed to me by John Hollerton, a senior spectroscopist at GlaxoSmithKline and co-author of the recent book Essential Practical NMR for Organic Chemistry. John intended it as a comment on a previous entry but I find it outstanding on its own and think that it actually opens a new thread.
Hence, here it goes:
Dear Stan,
I enjoy reading your blog which is often thought-provoking and always informative.
I am responding to your article "Am I drilling a hole in the middle?".
I have been chasing the ball of automated structural verification (ASV) for the last 20 years, sometimes intensely, sometimes from a distance. I am now (in)famous for stating that it will be a solved problem in "3-5 years". I have been saying this for the last 20 years. I believe it will happen but I don't think that it is a simple matter of AI. It may be a complex matter of AI but certainly not simple. A few years ago, I spent some time analysing what I and my NMR colleagues do when looking at NMR spectra. My conclusion was that it was a process of iterative hypothesis and test. This hypothesis generation is based on both NMR knowledge, non-linear data extraction (by which, I mean a certain degree of fuzziness in interpreting multiplicity, integrals, etc. which I don't have the space to go into here), chemical feasibility and the track record of the chemist who has synthesised the material(!). There are all sorts of other processes that go into hypothesis generation and these vary with the amount of experience of the NMR spectroscopist. The whole scope of information goes beyond the realm of simple (or even complex) NMR rules. It will be interesting to see if a computer ever matches that. Maybe in the next 3-5 years ...
So now I come to the purpose of ASV. I don't know of many (any) companies employing people to look at 50 spectra a day (except for specific, one-off projects). When I started working in NMR (now over 30 years ago) we looked at every sample that the chemists made. This worked out to be about 20-30 spectra per spectroscopist per day. Every day. This gave us a huge understanding of chemical shift, NMR phenomena, chemistry quirks and possible alternative products from common reactions. These days, chemists don't look at their own spectra except when they recognise that they have a problem. Their level of NMR ability is highly variable but it is safe to say that none have had the exposure to NMR in all of its wonder and serendipity. This is the first challenge. How do they identify that there may be a problem? I hope that ASV may help here by alerting them to the possibility that the data doesn't fit their compound. The second reason for having ASV is that my generation of NMR spectroscopists are the last to have gone through the intensive interpretation of NMR data. We are all getting older and starting to retire. We need something to help with this loss of knowledge and ASV could help. Lastly, we rely heavily on using LCMS to QC compounds coming from outside and also checking our screening hits. Modern NMR systems are capable of running huge numbers of samples but we can't look at the data - ASV could play a major role here so we can verify structure in a better way than weighing it!
I fully support the work being done by different groups, including Mestrelab, ACD and Bruker (maybe others?). I'm looking forward to ASV being available and reliable in the next 3-5 years!
Best wishes, John
What can I add, apart from a thanks for your kind opening words, and a confirmation that I know what you mean by thought-provoking. I, too, sometimes steam over what somebody else wrote and disagree so strongly I can't sleep whole night! I am joking, of course :-)
I share all your points and am tempted to expand on them, illustrating in detail my own [programmer's] perspective. Perhaps I will eventually do so. For the moment, my only comment is that 3-5 years, in my opinion, should be more than sufficient for a very decent and usable product with a performance equivalent to that of a fresh chemistry graduate with an extra training in NMR. Beyond that, I always remind my colleagues that even such simple algorithms as baseline and phase correction are still debated - more than 40 years after the first papers on them appeared. A much more complex topic such as ASV can therefore easily keep developing for a century or more and, once it becomes available, its uses can progressively invade areas more ample than those you foresee.
Who knows, maybe the knowledge of old spectroscopists who have gained an intuitive understanding of both the technique and its applications will keep living in the C++ codes of the programmers rather than in the minds of young chemists. Which might be OK, provided they live somewhere and don't have to be laboriously rediscovered again and again.
Hence, please, all of you who really know this trade, write it down somewhere ... Do not worry if the description is incomplete; it is the way you talk about it, not necessarily the content as such, that is most informative for the programmer.
|
April 07, 2011

It's not bucks
but I feel rich
anyway.
| Celebrating the first Million
Today this website totalled 1 million visits so I feel like celebrating! This actually includes just the pages on which I have put my own built-in counter. When all the 640 website pages are counted, the total number of visits indicated by my provider's statistics are almost 2 million (1930000)! On the average, a visitor displays 2.75 pages, implying 23.4 separate server contacts and 12.1 opened files, and downloads 283 kBytes. The daily average is presently about 1800 visits per day (drops to 1000 on weekends) which, I confess, I find a bit surprising myself. About 360 of those daily visits are to this NMR "Blog".
In a sense, it is a responsibility because any blunder I might make - and, being human, I am bound to make some - gets multiplied a millionfold. So I have to be careful to keep at least the Science objectively correct which often requires supplements of studies well beyond my real knowledge. As the Italians say, the School never ends. Fortunately, I enjoy this and promise to keep it up. In any case,
thank you all for dropping by and keep coming back!
The next stop is two million visits and let us try and reach it in much less time than the six years it took to total the first one. After all, the turf is now smooth and compact.
|
March 22, 2011

| Educating NMR Educators
Marshall Werner of Lake Superior State University (Michigan, USA) draw my attention to the Bootcamp for NMR Educators they are organizing for May 23 & 24 of this month.
This is a unique initiative (another one coming close is the book Modern NMR Spectroscopy in Education by David Rovnyak and his subsequent work in that area). In general, education in Magnetic Resonance is thriving, with a broad range of resources available on the internet, as well as in printed form. Of the former, the best known are the two Josef Hornak's online books, Basics of NMR and Basics of MRI (understandably, Joe is much involved in the Bootcamp). However, there seems to be never enough of it. The educational pages of my own website, for example, are the ones most frequently downloaded - much more often than anything else. Conference organizers everywhere are also catching up with this trend: I have just returned from a Conference in Slovakia (MMCE 2011) which had a distinctly educational flavor (in the next entry I will share some glimpses of it with you).
The Bootcamp, though, is the first MR event organized specifically to educate the educators, testifying to the width and depth of educational needs in our field. There is still room for a few more participants and lectures so, if you are interested, give it a thought.
|
March 12, 2011

Javelin NMR
| Javelin NMR
Speaking about holes drilling and NMR, there are much more direct connections than those discussed below. Vista Clara Inc, a geophysical-NMR company in Washington State, has last year developed, tested and sold first units of their new Javelin system which, I think, is a truly revolutionary advance in NMR well logging and complements nicely their larger and more traditional GMR system.
Javelin is a complete time-domain NMR instrument. They actually have two different javelins: Javelin Micro and Javelin Slim. The former, for example, is fitted into a tube just 42.4 mm (1.67 inch) thick, 2.13 m (7 feet) long and weighs mere 11.3 kg (25 pounds). This includes a magnet which produces an annular field around a section of the javelin, shaped in a way to permit NMR measurements at Larmor frequency of 250 kHz to 300 kHz in a thin (2-3 mm) cylindrical shell with a radius of 140 mm (5.5 inches).
For more details, consult the Javelin brochure.

In NMR well-logging, the inside-out magnet-and-probe configuration is a classic, of course. The novelty consists in the fact that Javelin can be lowered into holes of only 2 inches in diameter, such as those drilled for local water prospecting. The result is a vertical profile of various "types" of water around the drill hole (the reduced Figure on the right comes from an October press article in Fast-Times). Needless to say, the instrument can be easily transported in a pick-up truck. The Company in fact offers it not just for sale, but also rents it and/or measures the profiles as a service.
If you are really interested in all the technical aspects of this kind of magnetic resonance, Vista Clara is looking for people and has job openings for an engineer and a scientist (see Job Postings on their site).
It is nice to see that there are continuous news from the industrial front of MR. Despite the crisis, novel initiatives seem to appear more frequently than a decade ago, confirming the lasting vigor of the discipline. I actually can't keep pace with it all - there are two more items in my records, waiting since months to get published :-(
|
March 8, 2011
COMMENTS: 8
| Am I drilling a hole in the middle?
The last entry was not intended to be about social aspects of anything, it was about the destiny of NMR hardware. I continue thinking that nothing is more obsolete than an obsolete piece of technology and that most of the NMR systems being prematurely phased out will never make it back to operation again; they will be replaced by something else.
But then there was the Manuel Perez' comment, plus Josef Dadok send me a link to an insightful article by Paul Krugman, plus I found out a fascinating reflection of 1935 by none the less than Erwin Schrödinger and it all combined into a kind of mystic revelation which, however, left me with a bitter taste in my mouth.
Schrödinger says that we all resemble the dog with pleading eyes and a wagging tail who seems to say: "Master, please, throw a ball for me to chase! I got no goal and I need one!" Only, in our case, the Master is the Nature (true scientists) or some Noble Human Endeavor (applied researchers and technologists), and the ball is a challenging task.
I certainly do feel like the Schrödinger's dog (as opposed to his more famous cat). Presently, the ball in question is the use of NMR spectra for ASV (Automatic Structure Verification). It is certainly an attractive task which only recently appeared on the horizon of things that could be achieved within a reasonable time. Hence, me in Italy and my peers in Spain (thanks to internet, we can easily delocalize) are trying to teach a computer software the rules and skills of chemists specialized in NMR spectroscopy.
The typical application area for such a product is drug discovery, meaning Pharmas. I was told that these Companies employ masses of chemists who have to interpret some 50 spectra a day, having only 8h/50 = 9:36 minutes per spectrum. Clearly, they don't wag their tails, nor wish to pursue more balls. They were reduced to the echelon of routine workers and they hope that the new software might give them a few minutes more time to maybe ponder again that strange spectral peak that looked like an interesting challenge. Illusion, of course, because their top CEO's hope that the software will make half of the chemists redundant, leaving the rest doing a spectrum every 4:48 minutes (guess who will prevail).
Another question: is it conceptually possible to take, say, ten average chemists, "extract" their knowledge and skills, and instill it into an Artificial Intelligence in such a way that the AI will eventually perform better than each of them? The answer is YES, no doubt about it! Plus, the AI will outperform also the programmers who created it (speak about a Golem!). It may be difficult to pull it off, but it is a fascinating task (my tail is wagging like hell). The secret is the synergistic accumulation of knowledge inside the AI to make it evolve like a living organism (aborting hundreds of unfit builds, of course). In the end, it will inevitably beat the performance of each of the chemists and, on top of it, it will never tire nor strike. It will not even wag a tail, because AI's don't do that! They just perform.
But if it works as expected, what will happen in the Pharmas? On this point, Paul Krugman is inexorable: a category of jobs will simply disappear. There will still be the armed guards at the entrance, and the technicians who load the samples onto instrument trays, and a few top NMR spectroscopists. But in the middle, there will be a gaping hole.
So, here I am: a globalized guy outsourced into Lombardy woods, reading musty chemical NMR tomes, typing code, wagging a tail and, while/by doing so, silently drilling a hole in the whole world's middle class. Amazing! It makes me feel dizzy!
And, irrationally, a bit like I were a bloody bastard.
COMMENTS:
9 Mar 2011: Santi Domingues, (Mestrelab Research)
Stan, I have a comment on the last sentence of your last blog post.
On this issue, you should learn from many other people in similar situations and apply to yourself the usual: 'If I did not do it, someone else would'. This rationalization is used effectively in all paths of life and this is undoubtedly the case on this issue, as there are already several groups trying. Of course, all these efforts are commercially driven, as the demand for such a system is being clearly articulated by every small molecule department in pharma, and whilst there is demand, there will be companies and groups with families to feed who will work on this. I wonder what would happen if enough analytical chemists got together and created a foundation to finance lack of effort on ASV systems! But I guess, like in everything else, progress is inexorable.
Santi.
9 Mar 2011: Stan
Hi Santi. The saying 'If I don't do it, someone else will' must be used with care (you know what it can mean in a war). But in this case you are right - I just wanted to stress a point.
On the other hand, there is underway a pesky social process due to the "informatics revolution" which is going to cause bigger upheavals than what happened in the wake of the "industrial revolution". I went to my generic doctor yesterday for a routine visit and some prescriptions. I had to wait in a queue for 1 hour before being handled in 5 minutes. Afterwards I realized that during those 5 minutes the doctor never removed his fingers from a keyboard! We could save a lot of money (fewer doctors, no queues) if we delegated the routine stuff to an online auto-doctor, functioning as a filter between the Users and any flesh-and-blood physicians. Technically, it is certainly feasible. But it would make redundant about 50000 generic doctors just in Italy (my estimate). And yet the Universities keep spewing out legions of medium-quality graduates, most of them ear-marked as future redundants. In principle, if a skill can be taught, it can be also coded - there is no escape from this simple fact which harbors big social troubles.
It's not my fault, but I don't see any solutions of this riddle, ...
9 Mar 2011: Jacob deFeijter, (HTS-110)
Hi Stan.
My view is that people often get flustered whenever AI is mentioned. As you describe it, it is simply embedding a set of rules in software. This is a relatively straight forward task from a programming perspective. Sometime ago I managed a software team and we decided we could automate the analysis and interpretation of DNA profiles. The biggest challenge was the scientists' scepticism that it could be done as "several PhD's have worked on this for years and their system doesn't work very well". It took years for the gatekeepers to accept that the system my team developed should be evaluated. They then demonstrated that its was faster and more accurate than other systems and published a paper about it.
In fact, having non-scientists developing the system is probably better as they focus on making good software, The scientists' input should of course be the rules they apply and the current process they use for interpretation. The other recommendation I make is that you provide the programmers the raw data. Any additional filtering, smoothing etc should be done within the 'new' software so that all the information is available for interpretation.
I think it is a fun project and shouldn't take long to get a working system going. The most time consuming part will be to develop a "slick" user interface that helps to cut the interpretation time (and allowing for user overrides) to 2 minutes! Of course it may well be the sequence of steps in the new process is somewhat different than that used for the manual process so optimal use is made of the available processing power.
Regards, Jacob
[Post script:] Further to my earlier message, here is some more information. The paper I mentioned is http://www.ncbi.nlm.nih.gov/pubmed/19083817.
The programmer now works for another organization where he developed a similar system as well as a forensic DNA Database which has been installed in over half a dozen labs in 2 countries.
I think you may find that the system you have in mind can be developed quicker, better, and at lower cost if you work with an organization like the ones that developed RapidCall. I'll be happy to introduce you if you like.
10 Mar 2011: Stan
Hi Jacob. Thanks for the advice and, especially, for making the development of an ASV software sound as a Sunday picnic - it gives me hope. However, you possibly missed the fact that I was not calling for help. There are several groups working on it, I am part of one of them, and we feel like we have a good roadmap and are progressing well, despite the fact that some of us are severely handicapped by being PhD's.
This thread is about "drilling holes in the middle" and your comment sounds to me like you are pretty good in that business :-)
Catching on the occasion: your colleague Tijs Robinson promised a year ago to send me a 200 MHz spectrum acquired on your cute cryogen-free supercon magnet. All there was to do was a set of shims - an old task that looks simpler to me than an ASV wizard - but I did not receive anything yet. Could you remind him that as soon as I get the spectrum, I will write an enthusiastic article about it?
11 Mar 2011: a Senior Scientist (name withheld)
Dear Stan,
I think your comment about Companies employing masses of chemists to interpret NMR spectra is no longer the case, at least in my experience. The top CEOs had already decided many years ago that a small army of spectroscopists or data interpreters is an unnecessary expense and hands-on/walk-up instruments have made data gathering a trivial task. These days the chemist who makes the compounds has access to the NMR and other analytical data. He/she then assigns the spectra and decides if the data supports the supposed structure. As you know the skills required to do the assignment varies greatly between individuals and sometimes a chemist will just count protons and/or carbons and, together with MS data, assume that a structure is correct. Not forgetting that the chemist is always under pressure to produce more. As spectroscopists in the Pharma industry we spend a lot of our time training and advising synthetic organic chemists of the skills necessary in correctly interpreting analytical data. To my mind the attraction of ASV is to assist the 'bench' chemist in having surety that the right compound has been prepared.
We spectroscopists' wag our tails when we find that a chemist has completely miss-assigned the data when something like an unexpected biological result or odd physical chemistry behaviour reveals an incorrect structure. When ASV is 'cracked' we will have to find other things to wag our tails at in order to survive.
11 Mar 2011: Stan
I have no direct experience of how "life in a Pharma" might feel - I just reported hearsay from possibly disgruntled individuals and, to make a point, stressed it a bit. I understand that on the human level every Pharma might be a unique planet. ASV and other tools will of course continue developing and this will in time affect the way people work. Which is fine, provided they continue to work. Hence I sure wish you all the luck with "finding other things to wag our tails at in order to survive".
12 Mar 2011: Manuel Perez
Just to add a couple of things to your article. Life on any given day in a medium size chemistry department will go something like this: "Chemists will perform a series of chemical reactions, and based on their proton NMR and LC/MS data they will decide what to do next". This analysis takes a few minutes, MS data is a quick yes/no, and NMR data tends to be very quick as well, that for a good chemist of course. If the average chemist performs 4-5 reactions per day, this will result in the same number of NMRs and MSs spectra. Carrying the number forward to the rest of the year and thinking of a department with 100 chemists, these will generate ~200.000 spectra (at 5 minutes per sample, sample prep, walk to to analytical lab, wait for results ... for our imaginary department of 100 chemists that would represent ~690 days of chemists time per year). Obviously not all of them would need verification, but I can see, like yourself, a future world where all will be verified, the NMR will sit on the chemist's bench ...
Just a clarification at this point, I am only talking about in tube NMR experiments, the situation for the analysis of parallel/combinatorial chemistry final compounds is a different kind of problem. And so is the analysis of the file of compounds or the HTS (high-throughput screening) files of big companies.
Now a question of percentages, from experience I can tell that the good chemists are right over 99% per cent of the times (just because nothing is 100% in this universe). Why so high this percentage, because they have learnt through many years of experience which the common problems are, and do know exactly when to look for help, and it is only at this point where the specialist will get involved. But these are few and are being "asked" to retire, most chemists, less experienced, and wanting to be very productive, will be wrong more often. I really don't have a feeling for a percentage, but let's be optimistic and say 5% of their samples are wrong. Working with the same numbers as above, that would mean ~60 compounds which are not what it says in the tin/per year.
At this point there is something I have not mentioned, out of all those compounds most of them are not final compounds, the average optimal synthetic route will have 6 steps. Mistakes early on in the synthesis are less punitive in terms of time, false positives/negative in biological assays and wrong SAR. But nonetheless, they are a source of wasted resources (in terms of chemicals/solvents) and time lost for the chemists. Here the analysis gets a bit more difficult and I have to rely on experience and say that the average chemist will produce 3-4 wrong compounds per year, the number is much higher when we talk companies with less resources, tighter deadlines and more stressed chemists. How important these compounds really are? Usually, very important, since they tend to be novel compounds or compounds very difficult to synthesize = lots of time invested.
I would like to talk also about auto assignment and improving predictions, but this email is already too long ...
Best wishes, Manuel
12 Mar 2011: Stan
Manuel, I am overwhelmed! This is a full size article on its own - and a very informative one. Just consider that to me HTS always meant high temperature superconductors. Once you write about assignments and predictions, let us make it a self-standing article.
|
February 28, 2011
COMMENTS: 3
A chat in 2025:
Hi John!
Hi Cindy, how nice!
John, may I borrow
the 700 MHz NMR?
Sure, but it's K.O.
The console fell out
of my pocket and
stopped working.
Ah, no problem, I
would use my own
console anyway.
Is yours OpenMR
compatible?
Of course it is,
are you kidding?
No problem then.
Thanks, I will pick
it up with my Fiat
at 5 pm Friday.
What are you up to?
I will take it up to
the Green Mountain
pastures. We work
on fresh sheep milk
metabolomics.
Nice, bring me some
cheese, please!
John, does it have
a 12V car adaptor?
It sure does, dear!
I would also like
to look into an
old dynamic NMR
problem of mine.
I need spectra at
100 and 700 MHz.
How long it takes
to change the field?
Oh, the specs say
15 minutes, but I
can do it in ten.
Excellent!
I will do it Sunday
night by campfire.
Romantic girl! How
you evaluate such
multi-frequency,
multi-rate data?
I use Mnova, it
does everything.
I see, good choice.
John, would you
help me to set up
the acquisition
parameters?
Sure, when you are
on-field, go on-line
and we will remote.
Great, John,
I appreciate that.
...
| Post-crisis NMR market and technology
Today, if you have enough technical knowledge to install it, you can buy yourself a well-kept 500 MHz NMR spectrometer for just US$ 25000! (March 21: SOLD!) Of course, you will get no warranty, but still - just the included probes have a list price which is several times higher. If, instead, you want a turn-key second-hand system, installed and with one year warranty, ask Jon Webb of MR Resources (Massachusetts). They deal in used NMR equipment since late 70's when they took over the stock of the ill-fated IBM Scientific Instruments venture and they have an amazing choice (more than what shows on their web page), including almost any kind of spectrometer up to 700 MHz. Jon hinted to me that this is in part due to shrinking Pharma Companies.
Which is no big secret. Sixteen months ago I had an opportunity to visit the Pfizer research facility in Sandwich, UK. At that time, I got the usual treatment Big Corporations reserve for lowly visitors, complete with all the security crap and agreeing that, should a bird drop a natural product on their premises, I would never tell anybody and not even dream about photographing it. So it was on for-your-eyes-only basis that I admired a battery of over 30 high-field NMR machines, a still mightier platoon of mass spectrometers, and an essentially infinite array of large HPLC's. Today, the facility is closing down and all that hardware is on the used-equipment market for the taking, I guess. Similar stories are being told about Astra-Zeneca, GlaxoSmithKlein, and many others. There are so many phased-out high-field NMR spectrometers right now that even just getting rid of them might be a problem.
How will this affect NMR is everybody's guess, of course.
Personally, in the spectroscopy area, I perceive these aspects:
(a) Effects on major producers:
Short-sighted Companies might think that the used-equipment clot is a marketing disaster, because it will push down prices and preempt new sales. But others will understand that this is time for strategic decisions and bet on innovation, which is no doubt the only winning strategy. From the technical point of view, the existing equipment is obsolete. This certainly applies to consoles (electronics designed 10 years ago) but also to the present cryomagnets, particularly considering the deepening helium supply crisis. All of which may yet turn out to be a good thing because it leaves a lot of room for innovation. If I were a Top Gun in a Top Corporation, I would bet on table-top consoles and table-top cryogen-free supercon magnets that can be run in a few minutes from zero up to whatever desired field and then switched off for the weekend.
(b) Effects on used-equipment markets:
Those educational Institutions and small chemical Companies which don't have enough dough for new stuff can now set up an NMR lab with very small investment. The equipment will not be new, but it will still deliver at least 10 years of good NMR performance. The problem is installation and maintenance, of course, for which a decent commercial support existed so far essentially only in the US. I never understood why used-NMR businesses never sprang up in Europe. Are we too snobbish around here? There are some timid signs of change, like the French RS2D company, plus a number of scattered individuals who have the know-how to do it on an occasional basis, but it is far less than what the availability of used hardware would command.
(c) Effects on NMR markets in 'developing' Countries:
In areas like China, India, and Central and South Americas, the situation seems to be even worse. And yet it appears to me that the most logical solution for the current excess of US and European NMR hardware is to recycle it in those Countries, thus creating new centers of competence, as well as a future market. It is the only solution that should make happy everybody - from manufacturers to new end-Users.
(d) Effects on minor Companies and start-ups:
There is clearly a huge opportunity here. Everybody must innovate since otherwise new products would have to compete with the used ones. And it is no secret that small and/or start-up Companies are more dynamic and prone to implement revolutionary innovations than Big Corporations. In the last few years we have seen several small Companies to emerge, so the process seems to be underway. What puzzles me a bit is that there are so far no local NMR spectroscopy companies in China, India, and Brazil where the perspective local market should be huge. China now produces whole-body MRI scanners as well as small table-top MRI and time-domain NMR, but no spectrometers and, as far as I know, there are no NMR spectroscopy ventures in India and Brazil.
(e) Effects on MR engineering:
I have often witnessed how technical innovation gets postponed by manufacturers, to the chagrin of the engineers, because the current stuff is still doing its job and producing revenue. Innovation is not a smooth process - it takes a significant market jolt to induce the quantum jump to the next engineering generation. Well, during the last year or so, NMR spectroscopy market certainly did receive a jolt. Let us hope that it was significant enough, so the engineers can start cheering. Those who were paying attention might have noticed a new wave of engineering openings among small MR companies. It's not casual.
COMMENTS:
1 Mar 2011: Manuel Perez
Dear Stan. I just read the latest post in your blog, and I must admit it hit a nerve with me. Not only because it saddens me all you seem to remember from your visit to Pfizer is negative; I did try to be a good host, but it seems security are more memorable. But also because the biggest effect is the human cost, over 2400 direct jobs, thousands more affected.
Only in Pfizer Sandwich 6 NMR spectroscopists are being made redundant, including myself, plus more in the US sites. The cost to analytical sciences, in human terms again, is even greater, with hundreds of jobs going. The number of affected future academic collaborations is difficult to put a number to, but it will be considerable.
Again, you made a very good analysis, but I think you did miss the main term in the equation, the human one.
Best wishes, Manuel
1 Mar 2011: Stan
Dear Manuel, touché. I accept your comment as a slap I merit. Personally, you were indeed an excellent host. What I ironically referred to was the entry-point treatment and the general attitude of large Corporations when it comes to guarding their presumably precious secrets, the ones they strenuously enforce until the whole thing breaks down, and then they cynically discard both the secrets and the employees. After all, during the visit, even you were worried about where we could go and what we could see.
You and your colleagues were hit hard and I am sorry about that, but I and my four colleagues were also had, don't you think? We spent time and resources on presentations to people who were later laid off. The respective losses are very different, but the principle is the same.
In any case, I understand that any "social analysis" glossing over the human destinies of those involved may be irritating and carry with it an element of injustice.
I apologize for that and assure you that it is totally unintentional. I am certainly aware that behind every prematurely redundant NMR spectrometer there are scientists and technicians who were made redundant as well and that is more bitter than the aspects I am addressing.
4 Mar 2011: Stan
An aspect of the developing glut in used NMR instruments was pointed out to me by a research center accountant: though they did not phase-out any instrument, it still amounts to a de-facto depreciation of the institution's inventory, a loss which could be formalized and entered in the books. This is an effect I did not think about.
|
February 26, 2011
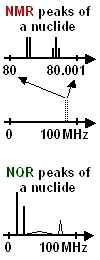
(all very schematic)
Change compound:
In NMR,
only the expansion
details change.
In NQR,
the whole spectral
pattern changes!
| An NQR picoPrimer
Terry Bayes (Roke Manor Research, UK) mailed me the following query:
Hi Stan, ...
I am interested in finding out if the NQR technique could be used to locate metals such as Zinc, and Manganese but although I can find data suggesting that they have high enough spin and nuclear quadrupole moments, I can't find any data on whether it is actually possible in real life and what frequency/power would need to be used. Have you come across anything that might give me a direction on this please? I am an electronic engineer, not a nuclear scientist, so simple is better! Thanks, Terry.
Terry, I am not an NQR specialist, so do not take my words as a distillate of truth, especially since the little I can say is not very promising for what you would like to do.
As I see it, there are two difficulties: (i) to start with, seeing a pure NQR signal under optimal lab conditions, and then (ii) exciting and detecting it remotely. By the way, 'pure' NQR means, in this context, without the application of an external magnetic field (NQR and NMR are often combined, but that is another story, and it does not change much what I am going to say, except that you also need a magnet). For reasons discussed below, one does not use any external electric field either (be careful: I have seen misleading descriptions of NQR on the Web, unleashed by some quantum-computing folks who have not done their homework).
The first problem has to do with the elusive nature of NQR signals.
Before we go into it, consider for a second a spin 1/2 nuclide (no quadrupole moment and no NQR, just NMR). When placed into a magnetic field, the Zeeman interaction makes it resonate at a Larmor frequency somewhere in an extremely narrow interval which is well known and nearly independent of the compound to which the nuclide belongs. In NMR, a resonance frequency deviation of 1% (10000 ppm) is unheard of and, usually, one can even estimate the expected peak's width and intensity.
Not so - not by a long shot - when the nuclide has a spin ≥1 and a large electric quadrupole moment! In pure NQR, the principal interaction is then the one between the nuclides' electric quadrupole moment eQ and the electric field gradient eq inside the molecular fragment of which the nuclide makes part. The electric field gradients are tensors centered at the locations of the nuclides and 'bound' to the local molecular environments - if the molecule tumbles, they tumble as well, if it stays fixed, they stay fixed. The tensors are traceless (I am sparing you the math) so if the molecule tumbles fast enough, the interaction averages to zero and there can be no pure NQR signal.
This is the first thing to remember: there is no NQR of liquids, nor of viscous systems nor, unfortunately, of biological tissues unless they are real dry (otherwise we would have nice 14N MRI options). The sample must be rigid enough to preclude rotational averaging.
Now, what about solids, such as the metals that interest you or maybe some [in]organic crystalline stuff. It so happens that the electric field gradients inside molecules (and metals) vary a lot from case to case. Consequently, depending upon its local chemical environment, the same nuclide can resonate anywhere between a few kHz and hundreds of MHz and there are no reliable prediction tools to tell us where (though experience does help a bit). You need a lot of patience to carry out the "fingerprinting" of each particular molecule of interest [1].
In case you wonder, we can't apply an external electric field gradient because we simply don't know how to produce it. Even a tiny fraction of a useful value would cause an electric breakdown (massive sparking) in any apparatus. The innards of molecules are the only places in the Universe where electric field gradients are strong enough to do NQR.
But this is not the end of the NQR blues. Spectral peaks do not have just position; they have also widths and, again, the variability of this parameter in pure NQR is enormous (hundreds of Hz to tens of MHz) when compared to what we are used to in NMR (fractions of Hz to tens of kHz). In particular, the width of a peak can be enormous, smearing it around until its height becomes smaller than the experimental noise and it can be no longer detected. Whether this will happen or not depends upon nuclear relaxation times which, in turn, depend on (a) the presence of unpaired electrons or (b) the presence of internal motions combined with (c) the magnitude of the interaction term (eq)(eQ). Hence, too big quadrupole moment and your chances to find a decent peak drop down (ditto for a too small one, obviously). Or take a strongly paramagnetic system (one width unpaired electrons) and your chances to detect a pure NQR peak become nearly null.
This partly explains why in metallic manganese no NQR signals has been observed at room temperature and what is going on is not quite clear. Metallic zinc (diamagnetic) should be simpler, but even there an NQR peak was seen only at 4.2 °K. All of which does not preclude the existence of zinc and manganese compounds with very nice NQR peaks. NQR is just not predictable enough! And you do not measure five samples a day, each maybe with 200 peaks, like in NMR; more likely, you study a couple of peaks for 5 years.
It also explains why nitrogen-14 (plus a few other nuclides) is so popular with the NQR crowd. Its quadrupole moment is just right, neither too big, nor too small, it is diamagnetic, it is present in many important organic compounds, and it is abundant in most solid explosives (hence the idea of using 14N NQR for remote detection of landmines and explosives [2,3]). When a 14N NQR peak is expected to exist, it is almost always found, though it can take time because its position is unpredictable.
Here we come to the second big problem. After decades of trying, nobody has managed to excite/detect NQR remotely [2,3,4]. By remote excitation/detection I mean something that decays with the square of distance and thus implies the presence of true electromagnetic radiation. The opposite is a near excitation/detection where signals tail-off with the cube of distance, implying 'just' a plain induction. There were great hopes once, but it was a wild goose hunt and all the hunters came back empty-handed. My impression is that at present hardly anybody still believes that remote NQR detection will ever work. Not even with such an NQR-friendly nuclide as 14N.
Which is a pity because of the enormous deminining problem [2]. On the other hand, I am afraid that should we find an efficient way to remove the landmines, they would just boost their production and deployment rates.
What is done in actual NQR practice [5-8] is a classical excitation and detection by induction coils positioned close to the object to be measured. Since, as explained, we can't interact directly with a nuclides' electric quadrupole moment, we use the standard NMR gear (minus the magnet) and interact, as usual, with its magnetic dipole moment, while between the RF pulses the dominant interaction is the one between the electric quadrupole moment and the internal, molecular electric field gradient. But the transmitter/receiver hardware and the sample need to be relatively close to each other (1 - 50 cm).
Ok, I do not want to say that this pico-primer is anywhere near to a real primer. But I hope that it will at least stop some of the quantum computing buffs from confusing their wishes with reality. As for you, Terry, if I were to invest into something, it would not be NQR detection of manganese-laced armor from the orbit. On the other hand, I could be wrong on all counts, of course, ...
References:
- Garroway A.N., Nuclear Quadrupole Resonance,
Figure K.2 is a nice example of NQR fingerprinting.
- Spin Resonance and Warfare,
Stan's NMR Blog (DOI), 01/15 (2008), and references therein.
- Spotting explosives and drugs: an Euromar 2008 update,
Stan's NMR Blog (DOI), 07/31 (2008).
- Daniels David J.,
EM Detection of Concealed Targets,
Wiley-IEEE Press 2009. ISBN 978-0470121696.
more >>
Chapter 4 deals with NQR detection of explosives.
- Sathyanarayana D.N.,
Introduction to Magnetic Resonance Spectroscopy ESR,NMR,NQR,
I.K. International Publishing House 2009. ISBN 978-9380026251.
more >>
- Parish Richard V.,
NMR,NQR,EPR and Mossbauer Spectroscopy in Inorganic Chemistry,
Ellis Horwood 1991. ISBN 978-0136255185.
more >>
- Straughan B.P., Walker S.,
Atomic, NMR, NQR, ESR and Mössbauer Spectroscopy,
Chapman and Hall, London 1976. ISBN 978-0470263235.
more >>
- Das T.P., Hahn E.L.,
NQR Spectroscopy,
Academic Press, New York 1958.
more >>
|
February 21, 2011
COMMENTS: 1

Richard Ernst

A pillory
| Bibliometric Discredibility Pillory
Yesterday I was browsing the Net for forthcoming MR events to list. One site I visited was that of the International ESR Society (IEPRS) which, alas, says that the place and date for the annual meeting of the Society to take place in 2011 will be announced later ...
While there, I have also checked out the EPR newsletter which the Society publishes. Not all its issues are free for non-members, but Issue 1 of Volume 20 is, and it carries a number of interesting historic sketches, plus a nice article by Richard Ernst entitled The Follies of Citation Indices and Academic Ranking Lists: A Brief Commentary to 'Bibliometrics as Weapons of Mass Citation' which, I think, everybody should read.
I could not agree more with what Richard says!
In 1970 I wrote an essay (Quo Vadis, Science) which, in some places, anticipates Richard's sentences. When I wrote it, I was young and naive, and the aberrant aspects of Science organization were less evident than today, but I must have anyway gotten them right if nowadays even Nobel Prize recipients are on the same wavelength! In my piece, I was blaming The Bomb (mistakenly, maybe, but let us wait until after World War III) and the massification of Science. Today I believe that it is primarily the mis-management of the latter that lead to the present citation craziness and related distortions.
In any case, thank you, Richard, now I feel less alone!
Regarding your exhortation to "establish an online Bibliometric Discredibility Pillory (BDP) and to encourage researchers to report there any abusers of bibliometric data", I am all for it and offer hereby my web domain and services to kick it off (though a separate domain will be better, I guess). Mail me the names of the first culprits and we can start.
Picture on the left: An abuser of bibliometric data exposed to public shame (I trust that he is not a Magnetic Resonance adept ... we don't do such things).
COMMENTS:
22 Feb 2011: Richard Ernst
Dear Stan, thanks for your moral support!
|
February 18, 2011

(BestAnimations)
| MR and metronomes
Since the annual March meeting of the APS (American Physics Society) is approaching (March 21-25, in Dallas, Texas), I have scanned its program to see whether anything relevant to magnetic resonance was being planned but, somewhat surprisingly, the search brought up only one interesting item.
Listed in Session 14 (New Ways of Communicating Physics) is a presentation with the hard-to-decode title Interactive NMR: A Simulation Based Teaching Tool for Fundamentals to Applications with Tangible Analogies whose Authors Sarah Griesse-Nascimento, Joshua Bridger, Keith Brown and Robert Westervelt, cited, "... created a module of eight interactive programs and accompanying lesson plans for teaching the fundamental concepts of Nuclear Magnetic Resonance (NMR) and Magnetic Resonance Imaging (MRI) ...".
The teaching approach is based on an analogy between spins and metronomes: "We begin with an analogy between nuclear spins and metronomes to start to build intuition about the dynamics of spins in a magnetic field. We continue to explain T1, T2, and pulse sequences with the metronome analogy". Which, I am sure, must be a great fun indeed.
On the other hand, I have tried myself quite a number of analogies to explain to neophytes various aspects of NMR, but none of them was flawless and some were outright unlucky (some analogies look cute but backfire in slippery ways). Which makes me curious about how the task has been handled in this case. I can't go to Dallas but, after the meeting, I will contact the Authors and see whether they might share with us any of their materials.
A side-note: In the Abstract, the Authors mention a "Magnetic Resonance Switch, a recent diagnostic technique based on NMR" which they also propose to explain by means of the metronome analogy. I confess my total ignorance about this technique (and so does Google) so that is another matter to be clarified (unless a reader helps me).
|
February 10, 2011
Forces on objects
around magnets

Top magnet:
cylindrical, with
an axial tunnel
Bottom magnet:
open-access,
horseshoe or
split-coil type
Blue arrows:
Magnetic field B
Red arrows:
accelerations
Small red circle:
FOV center
(force = 0)
Look at THIS!,
and check out
F.G.Shellock's
website
| More on MRI Safety
Most MRI accidents are due to ferromagnetic objects yanked from somebody's hands and, in the worst and sometimes fatal cases, slammed against a patient inside the scanner. Typical scenarios might involve a nurse approaching the scanner with scissors in her pocket, or a technician walking by the magnet with a heavy screwdriver in hand.
The nurse and the technician were probably both warned about the risk (if not, then there is a management problem). Perhaps they just forgot about the ferromagnetic object they were carrying - and there are technical and operational means to prevent this. But it might be that they believed they were smart enough to handle it (there is nothing to prevent stupidity). Maybe they were in the room with their tools on prior occasions and, since nothing much happened, they thought that the scary stories were exaggerated. In which case they did not appreciate how counter-intuitive can be the magnetic forces on objects. You can have an object snatched away from you just when you started feeling that all was going well - and you remain there open-mouthed and wondering what happened. Those who experienced it always say that the object suddenly became alive and they instinctively let it go.
There is an explanation for this which is at the same time physical and psychological.
Let me try to explain what I mean using the barest minimum of math.
Let us start with the physics. When a ferromagnetic object is placed into a magnetic field B, it gets magnetized. The magnetization M (a vector) of each of its voxels is oriented along B and its magnitude is Ms, the saturation magnetization of the material (bold letters indicate vectors, while normal letters denote scalars and vector magnitudes). Actually, in weak enough fields, M is an initially linear function of B, but this happens only when B is too small to exerts dangerous forces on the object. We will therefore consider only the situations when the magnetization is fully saturated and constant.
The energy of an object of mass W and a magnetization Ms aligned along a field B is
E = -W Ms B
The force F acting upon the object is given by the gradient of E,
F = -grad(E) = W Ms grad(B),
and it imposes on the object the acceleration
a = F/W = Ms grad(B).
Ferromagnetic objects are therefore pulled in the direction of the field gradient, the one in which the magnitude of B increases most rapidly, and they receive an acceleration which is proportional to the field gradient value and to their saturation magnetization, but independent of their mass. This has many interesting consequences.
Consider, for example, a cylindrical MRI magnet. Far outside, B and grad(B) are both nearly zero and the force is negligible. At the center of the magnet, inside the field of view area (FOV), the value of B is very large but, since MR requires a very homogeneous field across the FOV, grad(B) is null. This means that once the object is inside the FOV, it is no longer subject to any force. The centripetal pull on the object is really large only around the two ends of the magnet cylinder. If released somewhere in that area, objects start accelerating towards the center of the magnet and, once inside, they continue to fly by inertia.
The surprise factor is due in part to the invisibility of the magnetic field. But only in part!
Consider again a cylindrical magnet and think about it as a large dipole. Then the magnetic field B on its outside drops approximately with the third power of the distance d from the center, and the force on a ferromagnetic object is more or less proportional to 1/d^4 (the extra power being due to the grad operation). The problem is that our senses are not accustomed to something that varies with the fourth power of a distance! We are trained to handle forces which are either constant (such as weights) or vary linearly with a distance (elastic things). This means that our automatic reflexes are tuned for inverse powers from 0 to a bit over 1, but unprepared to handle powers as high as 4. Hence the instinctive panic when an elongated object suddenly comes to life in one's hands and starts pulling and twisting as it tries to align with the field gradient and fly away.
There is another interesting aspect which regards open-access magnets and which, I believe, has not been pointed out yet. Open-access magnets have large, flat poles and one can show that the strongest gradients around them point towards the edges of the poles, not towards the FOV! This implies that objects snatched by such magnets are likely to bang against a pole edge rather than against the patient. Open access magnets can not deliver as intense magnetic fields as the cylindrical ones but, from the present point of view, they are much safer.
To conclude, I think that personnel safety training sessions should be complemented with access to a true magnet (a scaled-down ferrite cylinder, perhaps) where the trainees could experience the weird behavior of small hand-held ferromagnetic objects inside strong and extensive magnetic fields. To experience is to believe!
|
February 6, 2011

More MRI Books
| 2011 MRI Safety Handbook
This year issue of
The Reference Manual for Magnetic Resonance Safety, Implants, and Devices,
published by the Biomedical Research Publishing Group, has been just released. It appeared on Amazon and, due to strong demand, was almost instantly sold out (fortunately, the situation is temporary and orders continue to be accepted).
This 'bible' of MRI safety, edited by Frank Shellock, is a must in every MRI facility. It is presently in its fifth update/revision cycle (previous editions:
2009,
2008,
2007,
2006).
There are five basic categories of health hazards associated with MRI facilities:
1) Strong main magnetic field (possible malfunctioning of electronic devices)
2) Static magnetic field gradients (mechanical forces on magnetizable objects)
3) Variable magnetic field gradients (electric currents induced in body tissues)
4) Radiofrequency pulses (RF burns; violations of EMI guidelines)
5) Contrast agents (safety issues common to all dangerous drugs)
Though these dangers have been extensively studied, understood and controlled, occasional accidents - even mortal ones - still happen, due invariably to human errors such as lack of attention and/or discipline.
Companies addressing MRI safety problems are listed among these (search for 'safety').
|
February 4, 2011
| NMR jobs and other news from New Zealand
Andrew Coy of Magritek averts me that they are trying to fill at least two engineering job positions, one of which is specifically labelled as NMR engineer. If I were younger, I would go for it myself since Magritek is a fine Company and New Zealand is a nice Country (a minor paradox: though they got lots of sheep, they import all pecorino cheese from Italy !?)
I also notice that Sir Paul Callaghan has been named the New Zealander of the Year 2011. Paul is a founder of Magritek and, in my opinion, he is also one of the ten best MR experts in the world. I just counted that his name appears on this website 28 times, so imagine what it must be worldwide. There is nothing I can add to the well written news article, except that I underwrite every word of it and wish Paul all the best in his present struggles.
Note: I wonder why we can't have an "Italian of the Year" and a "Czech of the Year" and, were there such Awards, who might get them? Suspecting what the answers might be, thumbs up for New Zealand. Well done!
|
January 29, 2011
COMMENTS: 2
| Keep your spinners smooth and clean!
The following spectrum (Fig.1) came recently my way via friends. The folder is marked "santonin" which, I guess, is a molecule dear to pharma chemists. Technically, the spectrum looks nice, at least at first glance: good resolution (with no apodization, the mean linewidth is < 0.7 Hz, while TMS linewidth is 0.3 Hz) and there are enough data points (57550, zero-filled to 128 K). An operator's comment says that it is a "single scan" spectrum while the spectrometer reports 16 scans; which is puzzling, but I have yet to see a dataset void of contradictions. Overall, I find it useful for testing the algorithms I am working on.


Fig.2 shows a piece of what I get after applying GSD (Global Spectrum Deconvolution, see [1], [2]). Quite satisfactory, but it took GSD an anomalously long time to locate and fit all the detectable peaks in this spectrum (about 30 seconds on my laptop). Wondering what was going on, I noticed that the list of peaks generated by GSD contained 939 of them - far too many for a molecule this size and complexity. Even considering that the S/N is good enough for all the first-rank 13C isotopomers to show up, I would not expect more than 200 peaks to get resolved. What's really going on becomes clear when a few strong multiplets in the spectrum are expanded vertically in order to see what happens around them at about 0.1% intensity level (Fig.3). In particular, look at the chloroform peak area in Fig.4.


There are tens and tens of badly phased weak "peaks" around every strong one! They look almost as noise but actually they are much stronger than the true noise. The latter is properly estimated by the system and, consequently, the old faithful GSD is trying hard (and with considerable success) to detect and fit all of the weak pseudo-peaks! Fortunately, I did enough lab work once to know exactly what the problem is (though I am still undecided whether and how to handle it in the context of my present work).
The culprit is simply a dirty spinner, nothing more scientific than that!
One can see the +-20 Hz sidebands around the strong peaks, so the sample was spinned and the spinning rate was the recommended 20 cycles/s. But the spinner (or the turbine) was dirty and therefore the rotation was not smooth - while spinning, the sample wobbled, trembled, and nutated, generating all kinds of messy rotational sidebands. One telltale sign of this happening is in fact the anomalous pseudo-noise around strong peaks, tapering off inversely with the distance from the main peak. Another indication, also confirmed in this spectrum, is that narrow peaks get affected much more than broader peaks. If this spectrum were not so well resolved, nobody would notice anything. And, naturally, no problem of this type can occur when you do not spin the sample at all.
It is often amazingly simple to cure this problem: a single swipe of the spinner with a soft cloth impregnated by isopropyl alcohol can work wonders. Sometimes, however, it may be the spinner turbine that has some dust particles sticking to its surface. That is more difficult to cure since one should get the turbine out of the magnet to clean it. In this case, it is best to enquire with the manufacturer about how to do it (unless it is in the manual). Needless to say, if your spinner is scratched, the consequences are very similar to when it is dirty, except that there is hardly anything you can do but replace it.
If you do not know where to buy isopropyl alcohol, this link is an option. The 70% stuff available from Amazon by the gallon is even better than the 99% pure one, since it removes also salty residues, not just greasy dust. It is also great for keyboards and the like.
WARNING: Never use acetone (!!!), nor chloroform (!!!), nor ethanol (!)
COMMENTS:
31 Jan 2011: Glenn Facey, (Uni Ottawa NMR Facility)
Hi Stan, Do you think it may be possible that some of these artifacts may be due to floor vibrations as seen here? I have also seen similar artifacts on Bruker spectrometers when the digital lock parameters are not set properly. Cheers Glenn
PS. I wish I could be in Italy now. It is so cold here (-24 C now with a wind chill factor of -30C) that one's nose freezes together when breathing.
2 Feb 2011: Stan
Certainly yes; vibrations can lead to very similar effects. One can understand what is going on by trying to link the problem either to a particular spinner (do other spinners give clean spectra?), or to a particular period (when did the problem start?), or to a particular location (try the spinner on a machine in another building). Once I fought with vibrations on the fourth floor of a concrete building where there was a production running on floors 1 and 2; the vibrations were so strong to generate a standing-waves pattern on the surface of a bowl with water (a simple vibration detector). Anti-vibration pads under the magnet did not help and eventually the installation had to be moved elsewhere.
Sometimes vibrations can cause artifacts similar to those described above even without rotation, but they must be strong or in mechanical resonance with the probe. In any case, one should try and vary the spinning rate. If the artifacts' envelope narrows when spinning at 10 Hz instead of 20 Hz, the problem should be due to the rotation.
Lock can cause a different kind of artifacts. There are two kinds of lock malfunctioning: One is random wandering around the correct value (large lock noise) due to insufficient signal or to saturation. These deform the peakshapes, but in a rather smooth way, not generating so many tiny peaks. The other possibility is a self-oscillation of the lock loop due to excessive feedback gain. In this case all peaks (even the small ones) get "smeared over" and apparently split into unresolved or barely resolved doublets (or even more complex multiplets). Also, while bad spinning or vibrations do not preclude achieving a narrow half-height width, lock problems usually make it all but impossible.
Regarding the PS: I understand - I stayed once in Saskatoon when it was minus 40 C! Here in North Italy we got this winter a -12 C dip one night, but now it starts looking like Spring. Why don't you move here?
|
January 27, 2011
Buzzwords:
TOTAL LOGGING
| Got memory, will log
Today I went to the neighborhood stationary shop which sidelines in computers and walked away with a 2 Tera-Bytes (!) USB hard disk. I have no real need for it, but it stays in the big pocket of my winter jacket and it costs just 100 eu-bucks! Meaning that the producer is getting just about 20 euros for it (5:1 is the canonic ratio in this trade).
It probably takes somebody my age to properly understand why I bought the damn thing.
As a programmer, I used to simulate coupled spin systems in early 60's on a machine that had a magnetic drum with 8 K words (they were 40-bit words, but still ...). Later I had to squeeze 7-spin simulations into the basic ASPECT computer configuration (Bruker silicon plus mine and Jürg Vogt's PANIC program) which consisted of 16 Kwords (24-bits/word, 8 K for OS+application, 8 K for data). But before that there was also a time when I was selling first FT-NMR spectrometers with woven ferrite-core memory blocks. The latter looked and felt like three 5x5 inch rectangles of heavy cloth (brocade-like), fixed into frames with a total capacity of 4 Kwords (12 bits/word for a total of 12 KBytes and 96 K tiny ferrite rings). I was selling them for about 8000 Deutsche marks/block, telling customers that "in NMR, they will never need to store more than 4 K data points". That was in 1976 and, given the prices, customers agreed. I wrote a reminiscence about it (in Italian). In early 80's I did a lot of Z80 based NMR. With that mythical micro I got the first glimpse of what a memory-unconstrained life might look like: it was about as fast as the ASPECT and it could address 64 KBytes of RAM! Unbelievable!!! Then, of course, the PC's arrived and changed the world, but even those took time to develop. Onboard RAM was limited to 640 K (for applications) and I still remember my very first and very own "modern" hard disk (IBM) with a capacity of 10 MBytes (was it 26 years ago?). After years of memory-hungry programming with kilometers of punched paper ribbons and cards, magnetic tapes (music cassettes), and floppies (those had a long evolution, too), I held this unbelievable thing in my [single!] hand and thought:
How could I - or anybody else - pretend to EVER fill this much memory space ?!
It reminded me of the sci-fi novel Orbitsville of Bob Shaw, if you know what I mean.
This reflection regards also the RAM mounted on various hardware boards. The boards I helped to develop at Stelar were always short on RAM. For example, our very first pulser board (1983 vintage) had just 8 K linear pulser steps (no cycles), quite insufficient for a decent CPMG-detected multi-block relaxation experiment. The last single-chip NMR board I worked on in early 2000's had an FPGA pulser with nested cycles, so that worry (almost) disappeared. But the board's acquisition RAM was just 1 MByte, amounting to 128 K of complex 32-bit words - good enough until you start with fancy arrayed and 2D experiments (there is still a list of experiments that I would like to try but no hardware, be it Bruker or Agilent or you-name-it, will let me to).
Today you can buy 199 US$ FPGA development boards which mount 64 MBytes RAM and, if need be, can stream data to a disk at incredible speeds. If an NMR manufacturer tells you that their machine can not do this or that because of memory capacity limits, it means that they are incredibly obsolete (incredibly meaning maybe 3 years).
So here I am with a 2 TBytes disk in my pocket and not a slightest idea about what to fill it with. If I can afford doing such a foolish thing, a medium-sized company should find it easy to arrange a memory bank of 1 PByte and a large Organization (stationed at Langley, VA, for example) a storage of 1 EByte (see the full list of SI prefixes). Moreover, they predict a tenfold increase in memory capacities (at constant prices) within this decade and a definitive move to solid state, with no moving parts at all.
Within the context of this blog, the question is: what does it mean for Magnetic Resonance?
I think that it brings up so many new possibilities to easily fill a book with them. Above all, it will probably bring about a radical change in the pilosophy of data acquisition (logging) and successive evaluation. In principle, the definitive goal should be to sample ALL front-end inputs and outputs such as the observe and lock RF channels, decoupler coil, gradient waveforms, power supplies, environment noise, you name it. They should be sampled continuously, synchroneously, and at high rates (up to giga-samples per second, whereever needed). Evaluation should be done later (later meaning anything from 1 ms to a century), but everything that happened in/around the instrument from the moment you inserted the sample (or the patient) to the moment you took it (or him/her) out should be recorded in the slightest detail at a very high rate. And I do not mean just taking note of the parameter settings which, due to imperfect/faulty interfaces, need not correspond to what really happened. I mean acquisition of the actual digital and analog signals as close to the front end as possible. The gear to do such total logging is available and relatively cheap, really.
I estimate that in this way one could just about fill my new 2 TByte disk with a single session at the instrument. But then, why not? The cost is just 100 euros, retail! And the disk can be recycled once the case is done with ...
Note: The original title of this entry was "Got memory, will use it" but I changed it to make it even more reminiscent of the classical "Got car, will work" of classified ads when they were still paid by the character.
|
January 23, 2011

| Upcoming NMRCAVES Workshop
The Organizers of this year's SMASH Conference have announced that they are planning an interesting Workshop, nicknamed NMRCAVES (NMR Computer Assisted Verification and Elucidation Systems). The name of the popular SMASH meeting stands for Small Molecules are Still Hot, indicating that it is primarily concerned with molecules up to about 500 Daltons which are the bread-and-butter of pharmacceutical R&D. Consequently, it provides an ideal setting for testing the performance of automatic and/or computer-assisted structure verification and elucidation algorithms (given their complexity, I prefer to call them wizards or, given their often perfidious behavior, Artificial Intelligences).
The Workshop will be chaired by Antony Williams and organized as a kind of 'contest':
Four moderately extensive sets of NMR data and molecular structures, combined in non-trivial ways into distinct test categories, will be pitted against software systems developed by participating groups from both academic and industrial environments.
The goals of the initiative, as stated in the NMRCAVES entry of Antony's blog, are:
to test the ability of algorithms to correctly elucidate the skeletons of unknowns,
to review the state of computer-based structure verification and elucidation, and
to institute a public NMRCAVES benchmark
The latter point, in my opinion, could help to speed up the evolution of these sophisticated NMR wizards. Clearly, the benchmark will consist of a set of data, molecular structures, and associated verification & elucidation tasks, against which to test any future algorithms and wizards. However, no benchmark should remain a static object. In informatics and electronics, benchmarks are common and their maintenance is usually handled by broadly-based Special Interest Groups. As one with a vested interest in this specific field, I would certainly like to see the birth of such an NMRCAVES SIG and I hope that the NMRCAVES Workshop migh kick it off.
|
January 20, 2011

| A new NMR textbook for chemists
A new textbook Essential Practical NMR for Organic Chemistry (Wiley 2011, ISBN 978-0470710920), written by Stephen Richards and John Hollerton (GlaxoSmithKline R&D), has just hit the market. I have not read it (yet) but certainly will do so and maybe add a comment. I initially could not find a Table of Contents (it is not on Amazon and I missed it on the Wiley page) until a friend pointed out the TOC link to me (scroll the page :-)
Personally, I love this sentence copied from the product description: "..., whilst stressing the importance of extracting the maximum available information from the simple 1-D proton experiment and of using this to plan subsequent experiments". I could not agree more.
Highly recommended and reasonably priced!
|
January 13, 2011

(N)MR Blog
| Welcome to SPINSIGHTS
Now that the merging of Varian Inc into Agilent Technologies is completed, apparently successfully and with no lost jobs, Agilent has started a new e-Zine named SPINSIGHTS. It has presently three branches, the most important of which is probably the SPINSIGHTS Blog, reserved for contributions by Agilent employees, consultants and co-workers. Judging from the first (but already numerous) entries, its orientation is technical and scientific and its quality is excellent. Needles to say, I have added it to my list of MR blogs.
Comment added on 14 Jan 2011:
I have commented a SPINSIGHTS entry and the comment was accepted. So SPINSIGHTS is not a strictly company blog; the comments are open to general public. But it is moderated, meaning that one should refrain from using four-letter words.
|
January 12, 2011

ParaHyperpol TM
| ParaHyperpol TM generates and handles para-hydrogen
The importance of para-hydrogen as a means to produce strongly polarized spin states and thus boost NMR signal intensities is well known and could hardly be undervalued, especially considering the dwindling supplies of helium-3 and the exorbitant costs of both helium-3 and xenon-29. Even non-enriched xenon costs up to many hundreds of US$ per milliliter, while hydrogen is plentiful and its cost is negligible.
Sure, to transfer the strong internal polarization of para-H2 (externally, its total spin is zero) to other molecules and make it detectable, a hydrogenation reaction is required. But this is a well understood aspect which has been worked out in considerable detail during the last quarter of century. The hydrogenation (often a marginal reaction) can be done in two ways - either inside the sample (PASADENA type experiments) or outside, in a special cross-polarization chamber and under RF irradiation which permits also an optional polarization transfer to other kinds of nuclides (ALTADENA type experiments).
However, up to now there has been no commercially available generator of hydrogen with a high para-hydrogen content, much less one combined with a reaction chamber, a built-in magnet, and a compact broad-band NMR console for the ALTADENA type techniques. For this reason the news I have just received from Gianni Ferrante, the President of STELAR is exciting and most welcome for the whole MR community. Stelar is an Italian company best known, so far, as the only producer of Fast-Field-Cycling NMR relaxometers.
This is the Gianni's e-mail:
Dear colleagues,
we are pleased to inform you that a new NMR apparatus, ParaHyperpol, has been recently introduced with the objective to offer a complete platform for PHIP (Para-Hydrogen-Induced-Polarization) polarization applications.
The system was developed by Stelar in collaboration with Invento, a spin-off company of Stelar based at the Molecular Imaging Center of the University of Torino.
Please download the ParaHyperpol brochure and do not hesitate to contact us for any comments, questions or more details ...
Best regards, Gianni Ferrante
Gianni will run a Stelar booth at the next ENC meeting, no doubt oce again associated with an Italian Caffè. So, if you want to know more about ParaHyperpol, drop by for a mini-cup of [real] coffee and a chat. Otherwise, contact Stelar or Invento right away.
References and Notes:
- Bowers C. Russel, Weitkamp Daniel P.,
Transformation of symmetrization order to nuclear spin magnetization by chemical reaction and nuclear magnetic resonance,
Phys.Rev.Letters 57, 2645-2648 (1986). Doi: 10.1103/PhysRevLett.57.2645.
This is the PASADENA seminal paper.
The name comes from the Authors' affiliation to CalTech in Pasadena, California.
- Canet Daniel, Aroulanda Christie, Mutzenhardt Pierre, Aime Silvio, Gobetto Roberto, Reineri Francesca,
Para-hydrogen enrichment and hyperpolarization,
Concepts in Magnetic Resonance 28A, 321-330 (2006). Doi: 10.1002/cmr.a.20065.
A nice mini-review of the topic.
- A large collection of PHIP papers by Joachim Bargon et al.
Bargon's group in Bonn (Germany) pioneered para-H NMR in Europe already in 80's.
- A primer on ALTADENA from Alex Pines group.
- ALTADENA =
Adiabatic Longitudinal Transport and Dissociation Engenders Net Alignment
The acronym is a joke. Altadena is an area close to Pasadena in California.
Hence PASADENA is an "in situ" method, while ALTADENA is "off situ, but not far away".
|
January 10, 2011
COMMENTS: 1
In a high field
{I·D·J} = I·{D}·J
where
{ ... }
stands for
rotational
averaging
and
D ≈ (3rr - r2)/r5,
where
r is the vector
interconnecting
the nuclides
High field
for 1H means
> 0.005 T

| Rotating Molecules versus Rotating Spins
A reader (name withheld) wrote me today:
Why does my teacher say that in liquids the dipolar interactions average to zero because of the random tumbling of the molecules? The corresponding energy term in the spin Hamiltonian is proportional to I.D.J, where I and J are the spin vectors of the two interacting nuclei and D is the dipolar interaction tensor. Even conceding that D is traceless, the whole term is a scalar and therefore invariant under any rotation. How can a rotational invariant average to zero because of rotations! My teacher must be wrong and there must be a different reason why the spectra of liquids have such sharp lines compared to those of solids.
Thanks for raising this point which is often misunderstood by students, while the source of the misunderstanding is frequently overlooked by teachers. The answer is that your teacher is right, but he could have explained it better. The problem may look trivial but it is not. As a referee, I even had to shred an MRI paper because the Authors thought that when a blood flow in an artery took a bend, so did the spin vectors ...
The misunderstanding consists in the fact that while the molecules tumble, or are being made to rotate together with the whole sample, the spin vectors stay fixed. One might say that, as long as the external magnetic field is strong enough, the spins don't care about what the molecules are doing! Mathematically, this means that the term I.D.J is not subject to the rotational tumbling as a whole; instead, the spin vectors I and J stay fixed, while D rotates randomly with the molecule and averages to zero.
Why is this so? It is all related to the magnitudes of the various interactions. Suppose we are talking about organic molecules (with their strongly magnetic protons and methylene -CH2- proton pairs) in a field that corresponds to the proton Larmor frequency of a 300 MHz spectrometer. Then for protons the principal interactions at play, expressed in Hertz (you can multiply them by the Planck constant to recover the SI energy units), are:
| + 300 000 000 |
Zeeman interaction between the magnetic field B and the nuclides |
| ± 100 000 |
approximate range of dipolar interactions between closest nuclei |
| - 7 000 |
typical chemical screening; the range is, roughly, -20 to -32 ppm |
| ± 20 |
approximate range of the indirect (scalar) coupling constants (J's) |
| < 1 |
magnetic field variations across the sample (field inhomogeneity) |
Clearly, the main magnetic field is what utterly dominates the proton's 'life' inside the magnet, somewhat like Earth's gravity dominates our's. The other interactions are distant perturbations, somewhat like the effects of the Moon or Jupiter on our own locomotion. Whether and how the molecules tumble is something the proton spin could not care about less, even though the proton mass is carried around. From the spin's isolated point of view, the Universe is unidirectional and almost perfectly axially symmetric.
Another way of describing this is to say that while the tensor D is solidly attached to the molecule, the spins are solidly attached to the main field and nothing can move them away from it but a near-resonant RF pulse! For example, in an electromagnet where the field is perpendicular to the cylindrical sample axis, it is surely not sufficient to manually rotate the sample by 90 degrees to obtain a transversal nuclear magnetization and therefore a nice FID! Even if the sample were solid and all the molecules really did rotate by 90 degrees, the spin vectors would still point in the same direction as before. To us old-timers who did NMR on electromagnets this comes as an easy-to-verify notion, but I can understand the doubts of present-day unfortunate youngsters who just confer their samples to a hissing pneumatic system ...
Legend to the Figure on the left: The blue arrow indicates the main magnetic field. A tumbling molecule (the green form) with two atoms, their nuclides (dark dots) and spins (red arrows) is shown in three different positions. Despite the tumbling, the spins (and the magnetic moments) of the nuclides maintain their orientation in space. They are shown here as parallel to the field but no such claim is actually made (this aspect will be discussed in a future entry). What might be the orientation and time evolution of the spins, the point here is that it is independent from and unaffected by molecular motions (except for weak, second-order relaxation effects).
The situation could be a bit different in very weak magnetic fields. Since the second (and the fourth) entries in the above table are not proportional to the main field, if the latter were weak enough to make the Larmor frequency smaller than about 200 kHz, the push & pull on the proton spin vectors due to molecular rotations would become substantial and the averaging process would become mathematically more complex (a rigorous theory of many MR phenomena in this regime is in fact still incomplete). This is what is meant by the term high-field approximation. For protons, "high field" starts at about 5 mT (about 0.2 MHz) while for other nuclides it is even less.
COMMENTS:
27 Jan 2011: Alfred Ross, (Hoffmann LaRoche AG)
Dear Stan, in context with the above posting, I ask myself the following question since some time (Gedankenexperiment):
1) Put a sample in a strong magnetic field and wait some time to polarize spins
(longitudinal magnetisation is created)
2) Take the sample out of the magnet quickly
3) Rotate the sample by 90 degree (the magnetisation is rotated by 90 degrees)
4) Bring the sample back in the magnet (neglecting field inhomogeneity on its way)
5) Detect an FID without a 90 degree pulse
I would expect that this works .... Kind regards for any comment, Alfred
30 Jan 2011: Stan
This thing is tricky and related to what is known as adiabatic field switching - a little known MR phenomenon which merits a separate entry because of its increasing practical importance in the area of storage and transport of nuclear hyper-polarization.
The fact is that your experiment will not work. As long as the field is appreciably large, the direction of nuclear magnetization stays "locked" to it and this invalidates your step (3).
- In liquids, even the Earth magnetic field (50 μT) is sufficient to do the trick. After you return the sample back to the magnet you might find it still considerably polarized (provided T1 is long even at low fields), but once again oriented along the main magnetic field. To see the residual magnetization, you need to apply a 90 degree RF pulse. The principle is the usual: of all the Universe, the nuclear spins only "perceive" the magnetic field - they follow that one and do not care about what you do with the molecules - or what the molecules themselves do in the way of tumbling.
- In solids, the molecules usually don't tumble and local magnetic fields can be much stronger than the Earth field. Consequently, as you bring the sample out of the magnet, the nuclides orient themselves along those and, since the local fields do rotate with the sample, it might look as though here your experiment could work. However, as you move the sample back into the magnet, the field each spin perceives varies smoothly from the local one to that of the magnet and all the nuclear magnetizations again end up aligned with the main field. It works so well that it even recovers the randomization of spin directions inside a polycrystalline solid when it is out of the magnet! This was used in the 50's to measure relaxation dispersion profiles of samples with long T1's (the first reference being N.F.Ramsey, R.V.Pound, Phys.Rev. 77, p.278, 1950). I tried it myself at H.S.Gutowsky's lab around 1972, using a fluoride salt (when pure enough, many solid fluorides have 19F T1 in excess of one day). I would keep it in a magnet for a few days, then take it out and keep it on my desk for a day, then put it back, apply a pulse and see a nice FID (while without the starting polarization step, there was no detectble signal). What you do with the sample while it is out of the magnet (like drive it around in your car) does not matter, but you still need an RF pulse to see the FID.
In both liquids and solids, field inhomogeneity during the sample transport is irrelevant.
All this was known, experimentally tested, and theoretically understood already in the 50's, even though a good quantitative description is still missing. There is a way to produce RF-less FID's of liquid samples in FFC (fast-field-cycling) NMR relaxometers where one can drive the field fast enough to achieve non-adiabatic field-switching conditions. But even there you must know what to do in order to see the phenomenon. I have a few such experiments on my hard disk (or, if need be, can collect more) so when I get around to writing the full entry, I will show some.
|
January 6, 2011
COMMENTS: 3


WIN ONE !!!

Dry ethanol
picoSpin spectrum
with Mnova GSD

The same with
GSD-enhanced
resolution
| The tiniest NMR spectrometer ever
One of the most exciting NMR news of the last Autumn was the appearance on the scene of picoSpin LLC with its homonymous table-top NMR spectrometer.
Despite its amazingly light weight (3.5 kg) and size (a footprint of a letter-format sheet of paper), picoSpin 45 is a true 45 MHz spectrometer with an innovative permanent magnet (1.06 T) and a field inhomogeneity below 0.1 ppm (typically 3 Hz). That, by itself is already an achievement. During last two years we have seen miniature permanent magnets with resolution of about 0.24 ppm and even less (half-height of 0.15 ppm, but with rather platykurtic peaks). Now picoSpin breaks the 0.1 ppm barrier and rolls out sample spectra with very decent peak shapes void of anomalous feet. Congratulations to the guy (JP) who developed the magnet!
But there is more to say. When I label picoSpin as a 'true' spectrometer, I mean that it not only has sufficient resolution for chemical and pharmaceutical applications, but also that it comes complete with an NMR lock and with a full set of field shims - almost everything you can find on any high-field machine. Plus it has a very decent 90 degrees pulse (20 μs; B1 of 290 &mi;T) and, though it uses a capillary, sensitivity of 300:1 (single scan on water). Plus, it is an FT design based on very modern electronics, presenting itself as a black-box accessory for a PC (just what I am trying to hammer in since years). With its Ethernet interface it can be actually connected to a network and controlled remotely!
Very interesting and unconventional is the sample delivery.
This is what picoSpin President, John Price, told me about it:
I know this will take people some time to get used to. A fundamental feature of this machine is that it uses a 300 micron ID capillary. The capillary starts at a fitting on the front panel, goes into the magnet and through the RF coils, and then ends at another fitting on the front panel. It takes about 20 μl of fluid to fill the capillary from inlet to outlet, but the volume inside the RF coil is less than 1 μl. There are many ways to get a fluid sample into the capillary. In the lab we usually use 100 μl glass syringes and we put a syringe port on the inlet fitting. ... I am pretty sure that once people get used to it they will really like this way of delivering samples. You can draw a sample from a test tube with a syringe and squirt it into the spectrometer in about 15 seconds, and have a nice looking FID a few seconds after that. There are no disposables in normal operation. In most situations, you don't even have to flush the capillary - you just displace the previous sample with the next one and start acquiring data. And deuterated solvents are optional - you use them only when there are solvent proton lines that obscure the lines you are interested in.
All these characteristics make the instrument eminently suited to plug the gaping hole NMR community lived with for the last 40 years or so: the lack of a simple NMR instrument to sit in the corner of an organic synthesis lab (together with an IR and UV/VIS spectrometer) for fast tests of synthesis steps (a market worth thousands of instruments world-wide), as well as the lack of a low-cost, modern educational NMR system. Indeed, picoSpin with its $20000 tab seems to be designed on purpose for these two tasks.
If you are interested, contact picoSpin, of course, but be prepared for a delivery queue. John is right now busily working on boosting up their production capacity. The first substantial-size series of devices will roll out in time for ENC or even before, but most of them are already sold!
A couple more points are worth mentioning:
First, picoSpin and Mestrelab Research just announced that every picoSpin unit will come with a free one-year User license for the Mnova software package, giving picoSpin customers access to the latest state-of-the art NMR spectra evaluation capabilities.
The second point is a reminder of the fact that much of the basic NMR know-how (Hamiltonians, all kinds of effects, ...) was established between 1955 and 1965 on mere 30 MHz spectrometers. So if you feel like sneering at the low operating frequency value, better think twice! Of course, second order-effects are inevitably more ubiquitous at low fields, but that is what linked-in computers are for and what they did not have in the 50's and 60's.
COMMENTS:
18 Jan 2011: Stan
An interesting article (picoSpin and the Incredible Shrinking Lab) about picoSpin appeared recently on Depth-First, written by Rich Apodaca whose point is rooted in what I call study of innovation pathways. He classifies picoSpin as a market disruptor which I find very interesting while, at the same time, I have an impression that Rich does not quite grasp the magnitude and speed of the tsunami that is going to hit NMR in the next one to two decades. The factor he perhaps overlooks is that, considering the recent advances in electronics, most of today's MR technology is obsolete and overdue for a major overhaul. Should Bruker, Agilent and Jeol think that picoSpin is a negligible niche device, they might be in for some surprises, and not just because it evidently contains pretty advanced electronics. For example, I would hardly call the educational market a niche; it is presently small only because it was so far strangled by exorbitant prices and commercial neglect (covered, in the FT-NMR spectroscopy area, just by Anasazi). And that is just an example - the same applies to NMR in developing Countries, plus a long list of more or less novel applications I could easily set up.
The situation reminds me of the first steps towards personal computers: do some of you remember the days of the Z80 machines, such as the Radio Shack 1980 computer kits or the Sharp MZ80K model, a few years before the 'real' PC's started and 'disrupted' the IBM's troglo-market? I think that picoSpin is like that - one of the first harbingers in what might become a long list (John Price should better keep watching his shoulders). It is everybody's guess when the first table-top 300 MHz black-box NMR spectrometer will become available - a feat which is already within the reach of a tinkerer (I suspect that plans for it are right now spread over more than one drawing board :-).
My bet is 2020!
4 Feb 2011: Chuck Miller (picoSpin LLC)
Hi Stan ... just wanted to send along a quick note of possible interest to you. Today, the Edison Awards organization officially announced the finalists of the 2011 Edison Best New Product Awards. Apparently, the picoSpin-45 was selected as a finalist for the Science & Medical - Diagnostic Aids category! Pretty exciting recognition for the team at picoSpin!
5 Feb 2011: Stan
Congratulations! It reminds me of the fact that the 'smallest ESR spectrometer ever built' of Active Spectrum won an award of similar weight less than two years ago. I think that should picoSpin and Active Spectrum join forces, there would be a lot of synergy - I can certainly see both instruments sitting on the same bench in a chemical lab/class and reinforcing each other. Of course, I am a really clumsy matchmaker, so this is as far as I will ever go :-)
|
January 3, 2011

MMCE 2011
| Spinning in Central Europe: MMCE and Valtice NMR meetings
I rarely promote specific Conferences but there will be two in the generic area I come from and I want to promote those. One is the bi-annual MMCE 2011 (Magnetic Moments in Central Europe), to be held March 16-20 in Tatranská Lomnica, Slovakia, and the other is the annual NMR 2011 (ex Valtice NMR with a long history behind its shoulders) to be held May 1-4 in Valtice, Czech Republic. Both are nice, cozy meetings with around 100 participants - the type I like most because parallel session are not needed and there is ample time for pleasant discussions (see my glimpses of last MMCE and of 23rd Valtice NMR).
I hope to participate at both meetings, but so far I pledged to show up only at MMCE where I will present a talk entitled Do we really understand Magnetic Resonance? (see the abstract). It falls in line with the musings I had been through in the last years and, in a certain sense, it will be a continuation of my recent Santander talk. However, if you have a look at the conference program, you will see that it is very rich and includes many speakers who (unlike me) undeniably understand their stuff and their in-depth talks will have a high tutorial content. If you are tempted to come, you really should! High Tatra mountains are real nice, especially when you enjoy the view while discussing NMR and do not need to actually climb them! Just remember that the already extended deadline for abstracts submission is January 16.
|
January 1, 2011


These chips can run
three NMR's each!
Read more

Kazuyuki Takeda's
OpenCore NMR
| Opening up 2011
First, let me wish All the Best and lots of Spin Discoveries in 2011
to all readers of this "blog", and to casual visitors as well !
To me the coming year looks promisingly exciting. I sure have a lot to write about, and it is not just the enormous backlog of requests and questions I still have to answer. I am not in a good enough shape today to write coherently about Science, but I want to mention at least two items which will involve me directly:
- Launch of the OpenMR initiative
This is something I have in mind since a long time. I am presently working on the format and contents of this venture (a kind of Manifest). It will be a permanent online initiative aimed at developing open-access (GNU license) magnetic resonance firmware and control software and establishing guidelines for OpenMR compatible hardware. Above all, it will be a free educational forum with the goal to teach graduate/postgraduate students, start-up companies, and plain hackers and basement tinkerers how to build top-class digital NMR consoles for less than 1 kEuros (there is less electronics in an MR console than in a mobile phone). Actually, we will be teaching each other, following the footsteps of similar open-access initiatives like Linux, Scilab, OpenCV, R, OpenCores, etc. There is a number of small MR Companies which had covered part of the same terrain (for example, I was part of the development team at Stelar), but they did it in isolation and the sources (IP cores) of their FPGA firmware are confidential. Big Companies also did part of the road, of course, but they are secretive and their results are not always faultless. Encouragingly, there was also an Academic initiative - the OpenCore NMR of Kazuyuki Takeda - which confirmed that a single-chip NMR spectrometer was feasible. Dr.Takeda's OpenCore NMR sources are in public domain and I am happy that he has agreed to join OpenMR. Judging from the number of technical enquiries I have received last year, the interest in OpenMR should be enormous. But we will need a legal standing, a domain, a wiki, a forum, a minimum of supervision, a lot of things. If the Organizers of the next ENC meeting (April 10-15, Asilomar, California) give us a chance, I would like to kick-off OpenMR there (if not, I will at least set up a poster and invite all of you to stop by).
- Automatic Structure Verification (ASV)
It is hardly a secret that I work with Carlos Cobas, Felipe Seoane, and others at Mestrelab Research on an ASV module for the Mnova NMR software platform. It is also clear that a good, user-friendly ASV software is a major challenge, a remarkable piece of Artificial Intelligence, and a futuristic tool much desired by many chemists, especially (but not only) in industrial environments.
There is a generalized feeling that ASV is coming of age and that its performance will soon reach a level sufficient for a successful deployment. This, unfortunately, raises considerable commercial expectations and competitive rivalry between the various groups of developers working on it. I say unfortunately because I would personally prefer if it could continue to be a pure intellectual endeavour. A recent episode made me understand that those days are now over, but I will nevertheless try and enjoy it as much as possible.
|
|
Archive
| For a complete list of all entries since 2005, see the running INDEX
|
|
Visitor #

ADVERTISE with us
NMR, MRI, ESR, NQR
Companies
Societies
Centres & Groups
Journals & Blogs
References
Free Texts
History
Links
BOOKS Lists
MATH | SOFTWARE
PHYSICS | CHEMISTRY
ELECTRONICS | DSP
WWW | Patents & IP
SPECTROSCOPY
MRI | NMR | ESR
Instruments
ARTICLES
Scalar relaxation
Biography of F.C.Yu
Hebel-Slichter effect
MR Antenna Theorem
NMR Dead Time
One-Page MR Primer
K-space and MRI
S/N Perspectives
OTHER
SI Units
Dimensions
Physics Constants
Math Constants
Science Links

Support this site!
SHOP from here:
COMPUTERS:
Deals
Bestsellers
Accessories
Calculators
This page is
SPONSORED by:


Random offers:
|